Cancer Cells Get Mitochondrial From Immune Cells that Invade the Tumour: A Study of T Cell Ownership
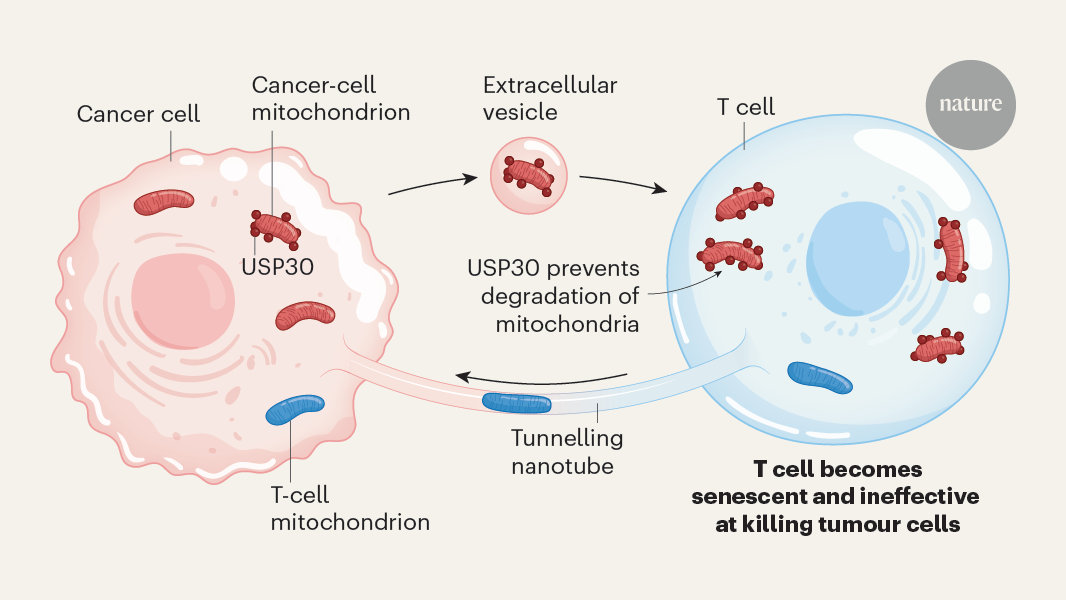
Most cells of humans and a number of other species have Mitochondria. Over the last two decades it has become clear that many types of cell can be converted to another type of cell. This process, called horizontal or intercellular mitochondria transfer, has been implicated in the maintenance of tissue health but can also contribute to cancer1. In a paper titled “Cancer cells get mitochondria from immune cells that invade the tumour”, it was shown that cancer cells get functional mitochondria from immune cells. In exchange, the cancer cells return mitochondria that harbour mutations and these organelles impair the T cells’ antitumour immune response. This bidirectional exchange of mitochondria enables cancer cells to support their metabolic needs while simultaneously creating favourable conditions for tumour growth.
J.R.B. is a member of the Scientific Advisory Board of LUCA Science, Inc., and has received research support from both Edgewise and Columbus Instruments in the past 12 months.
The idea of cellular ownership of the mitochondria has been challenged by this, according to the research team at the Washington University School of Medicine.
“My first thought was that this sounds crazy, like science fiction. But they seem to have the data for it,” says Holden Maecker, an immunologist at Stanford University in California, who was not involved in the research. This is a completely new biology that we weren’t looking at.
Tainted TILs were less able to divide and more likely to commit cell ‘suicide’, the team showed in cellular models. In mice with cancer, TILs that had imbibed alien mitochondria showed signs of T cell exhaustion — the loss of cancer-killing potential.
In vivo in vitro studies of the progression-free survival and overall survival of anti-PD-1 monoclonal antibody therapy in human tumours
consistent results were produced by all the repeated in vitro experiments. All in vivo mouse experiments were conducted with four to six mice per group and were repeated at least twice, which also produced consistent results.
The patient characteristics were compared between the two groups. The relationships between continuous variables between and among groups were compared using a t-test and one-way ANOVA, respectively. Tumour volume curves were compared using a two-way ANOVA. For multiple testing, Bonferroni correction was used. Progression-free survival and overall survival were defined as the time intervals from the initiation of anti-PD-1 monoclonal antibody therapy until the first observation of disease progression or death from any cause, and until death from any cause, respectively. The survival curves were analysed using a Kaplan–Meier method and compared with other groups. All tests were two-tailed with a predefined significance level of P < 0.05. The analyses were done using GraphPad software. The means and standard error of the means (error bars) are shown.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
T cell transfer models for the study of TIL04#9 cells in mice (B6 SCID) induced by tumour inoculation
TIL04#9 cells were labelled with MitoTracker Green. Twenty-four hours later, TIL09 cells were cocultured with either MEL05-MitoDsRed or without 10 mM NAC. The cells were stained with a LC3B-specific polyclonal Antibody and anAPC conjugated secondary Antibody, after which they were observed under a confocal laser. We used 10 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (Selleck Biotech) as a positive control.
We did the same thing by creating T cell transfer models using B6 mice. In brief, sorted CD8+ T cells (1 × 107) from splenocytes of C57BL/6J or OT-1 mice were transferred into the SCID mice 7 days after tumour inoculation (LLC/P29-OVA-MitoDsRed, 1 × 105; LLC/A11-OVA-MitoDsRed, 3 × 105). Tumours were collected 28 days after tumour inoculation to collect TILs for evaluation by flow cytometry.
All mice were maintained under specific pathogen-free conditions at the animal facility of the Institute of Biophysics (Chiba Cancer Center Research Institute and Okayama University). All mouse experiments were approved by the Animal Committee for Animal Experimentation of Chiba Cancer Center and Okayama University, and met the US Public Health Service Policy on Humane Care and Use of Laboratory Animals. When the maximum tumour diameter exceeded 20 mm, mice were killed as a humane end point. The supplementary fig 3 summarizes the experimental schematics.
A group of mice were purchased from Japan. C57BL/6J- Prkdc
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
In Situ Evaluation Of Cellular Proliferation: Flow Cytometry, Oxygen Consumption, Acidification, and ATP Production
The in situ cellular proliferation was evaluated with the IncuCyte ZOOM System every 6 h for 48H after cells were passed on 96-well plates.
Apoptosis was evaluated by combining Annexin V (Thermo Fisher Scientific) and ebioscience is fixable. Dye eFluor (Thermo Fisher Scientific) for live/dead cell staining. According to the manufacturer’s instructions, each cell was incubated with Annexin V and eFluor for 15 min at room temperature and then analysed by flow cytometry. Cellular proliferation was assessed on the basis of the dilution of cells labelled with carboxyfluorescein succinimidyl ester (CFSE) using a CFSE Cell Proliferation kit (Thermo Fisher Scientific) and flow cytometry. Cells were incubated with 10 μM CFSE for 20 min at 37 °C in 5% CO2, washed 3 times with RPMI medium and incubated for 3 days, followed by additional live/dead cell staining and flow cytometry analysis.
The kit was used for the assessment of cellular senescence. In brief, cells were incubated with bafilomycin A1 for 1 h at 37 °C in 5% CO2, then incubated with SPiDER-β-Gal for 30 min and analysed by flow cytometry.
The manufacturer’s instructions say that the analysis was performed using a flux analyser. The Seahorse RPM I medium contained 1 mM pyruvate, 2 mM glutamine and 10 mMglucose, which were followed by a series of miniplates. After equilibrated, the plate was put into an incubator and kept there for 40 minutes. The oxygen consumption rate was evaluated with sequential injections of oligomycin (1 μM), FCCP (0.75 μM) and rotenone–antimycin A (0.5 μM). The rate of acidification was measured by injections of sugars (10 mM, 1 M, and 2-deOXY-glucose 50 mM). The rate of production of ATP was determined using injections of otenone– antimycine A and oligomycin. The chemicals were purchased from a company. The data was normalized to the cell number.
Primary TILs were stained with 200 nM After 15 minutes, it is analysed by flow cytometry with MitoTracker Deep Red and Green. The membrane potential was evaluated on the basis of the ratio of each MFI4. Each Jurkat cell was stained with 250 nM TMRE and then analysed by flow cyclist.
The transfer of mitochondria from the cancer cells to the TILs was measured with or without 10 mM NAC. When using siRNAs, we cocultured the transfected cancer cells every 5 days with TILs. The manufacturer’s protocol says that siRNA transfection of cancer cells can be done using a reagent namedThermo Fisher Scientific. MEL03-MitoDsRed or MEL05-MitoDsRed cells were cultured in the presence of TILs for 2 days. Subsequently, TILs were analysed by flow cytometry.
The naive T cells were cocultured with MEL02-MitoDsRed Cells or MEL06-MItoDsRed Cells while they were being stimulated with an anti-CD3 monoclonal. Flow cytometry analysed the KLRG1 expression level and central memory fraction. Each CD8+T cell fraction is sorted by its effector memory and central memory.
Cells and TILs are checked for the status of mtDNA with capillary sequencing and several primers. The PrimeSTAR GXL DNA Polymerase (Takara) and the primers were prepared in 96-well plates. The cells from TILs were sorted into 96-well plates using a cell sorter. Oligonucleotides were amplified by PCR and sequenced (Eurofins Genomics).
MitoCheck Activity Assay kits were purchased from Cayman Chemical. The kit that was used for the Mitochondrial Isolation performed from cell lines was provided by the manufacturer. We used the activity buffer in place of the supplied mitochondria according to manufacturer instructions. Reactions were conducted at 25 °C using a FlexStation 3 microplate reader (Molecular Devices), with readings taken every 30 s for 15 min at the Central Research Laboratory of the Okayama University Medical School.
EVs in the cell culture medium were isolated using Total Exosome Isolation reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. In brief, 1 × 106 cells were cultured in RPMI1640 medium containing 10% EV-depleted FBS (System Biosciences) for 3 days. The conditioned medium was centrifuged at 400g for 5 min to remove cells, and the supernatant was centrifuged at 2,000g for 30 min to remove cell debris. The supernatant was passed through a syringe filter (0.45 μm pore; GVS North America) and then mixed with 0.5 volumes of Total Exosome Isolation reagent and incubated at 4 °C overnight. The mixture was then passed through a syringe filter (0.22 μm pore) and ultracentrifuged at 10,000g for 60 min at 4 °C. According to the manufacturer’s instructions, the pellet was resuspended in PBS, and then processed using a miniPURE- EV size-exclusion chromatography column. The resulting EVs were used in western blotting experiments. In brief, we used CD9 and TSG101 as EV markers70. To evaluate the purity of FBS-containing medium-derived EVs, we checked BSA71. We looked at the cytochrome c as a mus musculus.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Coculture of pBABE-puro TfR-ova and pVL-MitoDsRed cells with the USCS Xena database
We obtained RNA-sequencing expression data from The Cancer Genome Atlas (TCGA) in the Genomic Data Commons data portal of patients with melanoma from the USCS Xena database (https://xenabrowser.net). Expression data in tumours were used.
The cells were put into a culture dish and allowed to adhere after 24 h. Coculture with 1 106 TIL03#9 cells was started the next day. Twenty-four hours later, we began capturing images every 30 min using a digital holographic microscope (3D Cell Explorer CX-A, Nanolive). The analysis of the images was done using a software named “fiji”.
TIL04#9 and MEL04 cells were labelled with MitoTracker Green (Thermo Fisher Scientific) and MitoDsRed, respectively. 2 105 MEL04- MitoDsRed cells were cocultured with 1 106 TIL06#9 cells in a 35-mm glass-BOTTOM culture dish for 2 days and observed under a laser microscope. The cells were labelled with a antibody specific for CD45, clone HI100.
pcDNA3-TfR-OVA (Addgene, 64600)66, pBABE-puro (1764)67 and pVL-MitoDsRed (44386)68 were purchased from Addgene. The TfR-OVA cDNA was cloned into a pBABE-puro vector using In-Fusion Snap Assembly master mix (Takara Bio) according to the manufacturer’s instructions. The pBABE-puro-TfR- ova and pVsV-G genes were transfected with a reagent called Lipofectamine 3000. The pVL-MitoDsRed vector was transfected with packaging plasmids (pMDLg/pRRE, Addgene, 12251 (ref. 69); pRSV-Rev, Addgene, 12253 (ref. 69); and pMD2.G, Addgene, 12259) into packaging cells using Lipofectamine 3000 reagent. After 48h, the cells were transduced and concentrated into different cell lines. The cells were named MEL02.MItoDsRed, MEL05.MItoDsRed, MEL06.MItoDsRed and MELc03.MItoDsRed. The cell lines that were produced were called P29-OVA- MitoDsRed and A11-OVA- MitoDsRed.
The cell specimen was put in 0.1 M PBS and contained 2%glutaraldehyde and 2% paraformaldehyde for 16 to 18 h. Post-fixation was performed with 2% osmium tetroxide for 1.5 h. The specimens were dehydrated and embedded in a low-viscosity resins after washing with PBS. Subsequently, 80-nm-thick sections were prepared using an ultramicrotome (EM-UC7; Leica) and stained with uranyl acetate and lead citrate. The specimen was viewed using a transmission electron microscope. We had a number of cristae perchondrion.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Microorganisms used to grow and maintain mycoplasma-free cells in the environment of a microcavity incubator in the treatment of cancer cells
All cell lines were used after confirming that they were mycoplasma-free, which was assessed using a PCR Mycoplasma Detection kit (Takara) according to the manufacturer’s instructions.
To grow cancer cells, the digested tumours cells were cultured in a medium containing 10% FBS, 1% penicillin–strepomycin and 1% amphotericin B. Tumour cells were passaged at approximately 80–90% confluence and used when free of fibroblasts and proliferating beyond the tenth passage. The digests from the tumours were fed into the RPMI 1640 medium and put into a humidified 37C incubator. Half of the medium was taken out and replaced with fresh medium and rhIL-2 every 3 days.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Performance Assessment of Polymorphic Variants in Somatic Cancers Using the Allele Frequency Metric and the EAGLE Data Set
After pooling the EAGLE results from all samples, we used z scores determined from the variant allele frequency (VAF) per position as a metric to evaluate potential variants. Alterations with z scores > 3, VAF > 0.2 and read depths exceeding 100 were selected to minimize false positives from sequencing errors. In cohorts B, C1 and C2, variants with VAF > 0.85 were initially considered polymorphisms and excluded from final reporting under the assumption that somatic variants would not exhibit high VAFs owing to the presence of a substantial fraction of non-cancerous cells in the tumour tissue in the specimens collected. There were 10 hits in the final output and heteroplasmy levels were set to 0.85 so Haplogrep was used to identify possible haplogroups. Variants labelled as ‘hotspot’ and ‘local private variant’ were regarded as polymorphic variants; filtered variants were visually inspected to exclude probable sequencing errors at the termini of PCR amplicons or in homopolymer sequence stretches. In performance assessment, we compared the presence of true variant, confirmed through a combination of matched and normal tissue samples, with variant that were only from tumour samples. The true variant was called with a false negative rate of 12.2% and a false positive rate of zero.
The protocol for this study was approved by the appropriate institutional review boards and ethics committees of Yamanashi University Hospital, Chiba University Hospital, Shinshu University Hospital, Okayama University Hospital, Kindai University Hospital and Saitama Medical University International Medical Center. The study was done according to the principles of the Declaration of histieth.
Ten patients with melanoma, one with breast cancer and one with skin squamous cell carcinoma were enrolled in this study, and samples were used to establish TILs and matched cancer cell lines (cohort A; Supplementary Table 1). Yamanashi University Hospital was one of the hospitals where participants had undergone surgical resection. The tumour tissues were processed as previously described60. In brief, surgically resected samples were enzymatically digested with 0.1% collagenase, 0.01% hyaluronidase and 30 U ml–1 deoxyribonuclease (Sigma-Aldrich) in RPMI1640 (Thermo Fisher Scientific) at room temperature. The digested tumour cells were subjected to filtration and density-gradient separation before use. Blood mononuclear cells were obtained from donated blood. All participants provided written informed consent.



