Ultracold Immunoblot Analysis of the KBTBD4-PR Mutant PDX (ICB1572) using the LICOR Odyssey CLX Imaging System
KBTBD4-PR mutant PDX (ICB1572) tumour was freshly isolated from mouse cerebellum, dissociated to a single-cell suspension and plated at 1 × 106 cells per well in a six-well plate in Stem cell media. Cells were immediately dosed with the HDAC1/2 inhibitor RBC1HI or the NEDD8-activating enzyme inhibitor MLN4924 and incubated at 95% humidity and 5% CO2. 24 h later, cells were collected and lysed in the RIPA buffer. Immunoblot images were captured using the LICOR Odyssey CLX Imaging system.
Offline bRP fractionation was performed to separate peptides over a 96 min gradient with a flow rate of 1 ml min−1. 5 mM was the maximum amount of solvent A. ammonium formate/2% acetonitrile and solvent B was 5 mM ammonium formate/90% acetonitrile. In total, 96 fractions were concatenated into 24 fractions for proteome analysis. Then, 5 µg of peptides from each of the 24 fractions was transferred into HPLC vials, frozen and dried in a vacuum centrifuge for analysis. 1 g from each of the 24 fractions was injected for LC–MS/MS analysis.
In the acidic pH LC–MS/MS analysis, each fraction from basic pH LC was dried by a Speedvac and was run sequentially on a column (75 µm × 35 cm for the whole proteome, 50 µm × 30 cm for whole proteome, 1.9 µm of C18 resin from Dr. Maisch, 65 °C to reduce backpressure) interfaced with a Fusion mass spectrometer (Thermo Fisher) for the whole proteome where peptides were eluted by a 90 min gradient (buffer A: 0.2% formic acid, 5% DMSO; buffer B: buffer A plus 65% acetonitrile). The settings included theMS1 scan, a 60,000 resolution, and 50 ms maximal ion time, as well as 20 data-dependentMS2 scans.
The computation of identification and quantification was done with a search engine. All original target protein sequences were reversed to generate a decoy database that was concatenated to the target database. Putative peptide spectrum matches (PSMs) were filtered by mass accuracy and then grouped by precursor ion charge state and filtered by JUMP-based matching scores (Jscore and ΔJn) to reduce false discovery rate (FDR) below 1% for proteins during the whole-proteome analysis. If one peptide could be generated from multiple homologous proteins, on the basis of the rule of parsimony, the peptide was assigned to the canonical protein form in the manually curated SwissProt database. The highest PSM number was assigned the peptide to because there had been no definition of a valid form. We performed the analysis in the following steps, as previously reported, with modifications49: (1) extracting TMT reporter ion intensities of each PSM; (2) correcting the raw intensities on the basis of the isotopic distribution of each labelling reagent (for example, TMT126 generates 91.8%, 7.9% and 0.3% of 126, 127 and 128 m/z ions, respectively); (3) excluding PSMs of very low intensities (for example, minimum intensity of 1,000 and median intensity of 5,000); (4) removing sample loading bias by normalization with the trimmed median intensity of all PSMs; (5) calculating the mean-centred intensities across samples (for example, relative intensities between each sample and the mean); (6) summarizing protein or phosphopeptide relative intensities by averaging related PSMs; (7) finally, deriving protein or phosphopeptide absolute intensities by multiplying the relative intensities by the grand-mean of three most highly abundant PSMs. We also corrected the data using y1 ion-based technology. Supplementary Data 1 is included.
Differentially expressed proteins were identified using an empirical Bayes-moderated t-test to compare treatment groups with the limma R package (v.3.54.2)50. Low expressions were defined as the lower 25th percentile of the means of the protein expression, and proteins with a prevalence of low expression in more than 70% of the samples were filtered out. 7,731 out of 11,428 were retained for further analysis. The criteria for differential expression included a P value and fold- change greater than 1.5. The ggplot2 is a package used to create volcano plots. The R environment was used. PPI networks were constructed using STRINGdb (v.12)51, with a confidence threshold greater than 0.7. The networks were visualized using a software called Cytoscape. Interaction data were sourced from text mining, experiments and existing databases. Supplementary Data 1 and 2 can be found here.
HEK293T cells (Thermo Fisher) were a gift from B. E. Bernstein (Massachusetts General Hospital). Gesicle Producer 293T cells were a gift from D. R. Liu (Harvard University/Broad Institute) (Takara, catalogue no. 632617). K562 and CHLA-01-MED are from ATCC. All mammalian cell lines were cultured in a humidified 5% CO2 incubator at 37 °C and routinely tested for mycoplasma (Sigma-Aldrich). ICB1299, CHLA-01-MED, and MED411FH-TC cells were cultured in stem cell media (50% DMEM/Nutrient Mixture F12 (DMEM/F12) plus 50% Neurobasal-A Medium supplemented with B-27 supplement (without vitamin A), 1 × GlutaMAX (Invitrogen), 1 mmol l−1 sodium pyruvate (Invitrogen), 1 × MEM Non-Essential Amino Acids Solution (Invitrogen), 25 mmol l−1 HEPES, 20 ng ml−1 basic fibroblast growth factor and 20 ng ml−1 epidermal growth factor). ICB1299 cells were cultured in Matrigel-coated plates and CHLA-01-MED and MED411FH-TC cells were cultured in low-attachment plates. The HEK293F cells were obtained from a laboratory. RPMI1640 and DMEM were supplemented with 100 U ml−1 penicillin and 100 µg ml−1 streptomycin (Gibco) and FBS (Peak Serum). K562 cells were cultured in Gibco and supplemented with 10% FBS. The cells were cultured with 10% FBS in the DMEM. HEK293F cells were cultured in Freestyle 293 Expression Medium (Thermo Fisher) with shaking at 125 rpm. Spodoptera frugiperda (Sf9) insect cells (Expression Systems, catalogue no. 94-001F) were cultured in ESF921 media (Expression Systems) in a non-humidified and non-CO2 incubator at 27 °C with shaking at 140 rpm. The High Five and Expi Sf9 cells were purchased from the same company. B85502 and A35243, respectively), with Grace insect medium (Thermo Fisher, catalogue no. 11595030) supplemented with 10% FBS (Cytiva) and 1% penicillin-streptomycin (Gibco), and cultured at 26 °C. All cell lines were tested for mycoplasma and then verified by short tandem repeat profiling.
Engineered virus-like particles (eVLPs) were produced as previously described71. In brief, Gesicle Producer 293T cells were seeded into T-75 flasks (Corning) at a density of 5 × 106 cells per flask. After 20–24 h, a mixture of plasmids expressing VSV-G (400 ng), MMLVgag–pro–pol (3,375 ng), MMLVgag–3×NES–ABE8e (1,125 ng) and an sgRNA (4,400 ng) were co-transfected into each T-75 flask using jetPRIME transfection reagent (Polyplus) according to the manufacturer’s protocols. Then, 40–48 h after transfection, the producer cell supernatant was collected and centrifuged for 10 min at 4 °C and 2,000g to remove the cell debris. The clarified eVLP-containing supernatant was filtered through a 0.45 μm PVDF filter (Sigma-Aldrich). The supernatant was concentrated using a cushion of 20% (w/v) sucrose in PBS. Ultracentrifugation was performed at 26,000 rpm for 2 h at 4 °C using an SW28 rotor in an Optima XE-90 Ultracentrifuge (Beckman Coulter). After ultracentrifugation, eVLP pellets were resuspended in cold PBS (pH 7.4). eVLPs were kept at 80 C. eVLPs were frozen before use so that they would not get freeze–thaw.
The mutational library was cloned into pSMAL mCherry using Gibson assembly. The introduction of a restriction site called the BamHI was used in Supplementary Table 6 to prepare the backbone for the Gibson assembly. The correct linearized backbone was isolated using a gel electrophoresis technique and purified using a gel DNA Recovery Kit. Then, 190 ng of linearized vector and 13.15 ng of each sub pool were used for each Gibson reaction of 80 µl using HIFI DNA Assembly Master Mix (NEB). The Gibson reaction was incubated for 1 h at 50 °C and DNA was isolated by isopropanol precipitation and transformed into Lucigen Endura Competent Cells according to the manufacturer’s protocol. Cells were recovered in Lucigen Endura Recovery Media for 1 h at 30 °C and later plated and grown overnight at 30 °C. The collected colonies were used to extract the plasmid library. Purified sub pools were combined to make up the final library and sequence.
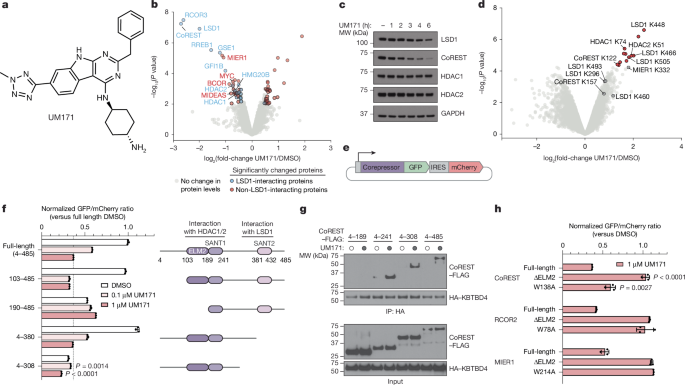
For fluorescent and stability reporter constructs, CoREST, MIER1, RCOR2 and KBTBD4 were cloned into Cilantro 2, a gift from B. Ebert (Addgene, 74450). The CoREST and HA–KBTBD4 constructs were cloned into pcDNA3. The mCherry ORF was introduced into pSMAL to create the coding sequence, which was a gift from J. E. Dick. For inducible expression constructs, KBTBD4 coding sequence (CDS) was cloned into pInducer20, a gift from S. Elledge (Addgene, 44012). A gift from T. Cech, pFastbac was used to clone two bacmid expression genes. The construct for structure determination was created with the assistance of a pFastBac sequence and 10 Histag and MBP tag at the N end. For eVLP constructs, sgRNA sequences were cloned into pU6-sgRNA (a gift from D. R. Liu) by PCR amplification, and co-transfected with pCMV-MMLVgag-3×NES-ABE8e (Addgene, 181751), pBS-CMV-gagpol (Addgene, 35614) and pCMV-VSV-G (Addgene no. 8454), gifts from D. R. Liu, P. Salmon, and B. Weinberg, respectively.
eVLPs were produced as previously described22. A total of 106 cells per flask were used to seed Gesicle Producer 293T cells. After 20–24 h, a mixture of plasmids expressing VSV-G (400 ng), MMLVgag–pro–pol (3,375 ng), MMLVgag–3xNES–ABE8e (1,125 ng) and an sgRNA (4,400 ng) were co-transfected into each T-75 flask using jetPRIME transfection reagent (Polyplus) according to the manufacturer’s protocols. At 40–48 h after transfection, producer cell supernatant was collected and centrifuged for 10 min at 4 °C and 2,000g to remove cell debris. The clarified eVLP-containing supernatant was filtered through a 0.45 μm PVDF filter (Sigma-Aldrich). The filtered supernatant was concentrated by ultracentrifugation using a cushion of 20% (w/v) sucrose (Sigma-Aldrich) in PBS. The procedure was carried out using aBeckman Coulter Ultracentrifuge and a SW28 rotor. After ultracentrifugation, eVLP pellets were resuspended in cold PBS (pH 7.4). eVLPs were frozen and kept at 80C. eVLPs were thawed on ice immediately before use and repeated freeze–thaw was avoided.
K562 cells were plated for transduction in 96-well plates and had a density of 50,000 cells per well. The culture medium was put directly into each well with BE-eVLPs. After 6 h, fresh medium was added, and after 48h another 100 l of medium was added. After 72 h, cellular genomic DNA was isolated and re-analyzed. Before degradation assays were performed, transduced cells were allowed to recover for 7–10 days.
ICB1299 and CHLA-01-MED were transduced with eVLPs, and cultured in Stem cell media. On day 3, the cells were collected for genotyping. Cell viability was assessed on day 4 and day 11 of the previous week, followed by day 3 and day 10 of the previous year. End point readings were normalized to that of reference to determine relative growth during 7 days of culture. For immunoblotting, ICB1299 cells were transduced with eVLPs and cultured in Stem cell media for 5 days before collecting for immunoblotting or genotyping. Supplementary Table 2 contains primer information for genotyping.
Genomic DNA was purified using the QIAamp DNA Blood Mini (Qiagen) or QuickExtract DNA Extraction Solution (Biosearch Technologies) according to the manufacturer’s protocol. We put 100 ng of DNA through a first round of Q5 hot-start high fidelity DNA (New England Biolabs) to amplify the interest and attach overhangs. The 2 l of each product was amplified in a second round of the polymerase chain reaction. The final amplicons were scopd on an Illumina MiSeq. The data was processed using parameters such asquantification window size 20 andquantification window center 10. 20 –exclude_bp_from_left 0 Excludingbp from right 0 was excluded. –min_average_read_quality 30 –n_processes 12 –base_editor_output.
A linker-FKBP12F36 V-2xHA-P2A-PuroR cassette was inserted into the C terminus of HDAC2 with Homology- directed repair. The C end of HDAC2 was targeted by sgRNA according to a manufacturer’s protocol. In brief, 2 × 106 cells were washed twice with PBS and resuspended in buffer R. The sgRNA and the repair vector (0.5 µg) were added to the cell suspension, and electroporated at 1,350 V with a 10 ms pulse width for three pulses using the Neon Transfection System 100 µl kit. After electroporation, cells were immediately transferred to prewarmed medium. Cells were selected with puromycin for 10 days and then single cell sorting was done on the Mo Flow Astrios EQ Cell Sorter. Single-cell clones were checked out by blotting and genotypic testing.
K562 KBTBD4-null CoREST–GFP cells were generated as described above. KBTBD4 overexpression constructs were cloned into pSMAL mCherry and point mutations were introduced into coding regions using standard PCR-based site-directed mutagenesis techniques. The over expression constructs which were carried by Lentiviral particles were used to transduce cells. After 48 h, cells were treated with 1 M UM171, or 0.12 fluid ounces per square inch. The percentage was measured for mCherry+cells in each condition and shown in supplementary fig. 1c.
HEK293T cells were transfected with 2 µg of pcDNA3 HA-KBTBD4 plasmid (mutant or WT) and with or without 3 µg of pcDNA3 CoREST–FLAG (full length or truncated) using PEI MAX transfection reagent (Polysciences) according to the manufacturer’s protocol. After 48 h, cells were treated with 1 M Mln4924 or 1 M UM171 and 10 M SAHA, or with 10 M CI-994. Cells were washed twice with cold PBS and flash-frozen. The ice in the lysis buffer made it difficult for cells to thaw. Tris-HCl has a pH of 7.5. The lysates were cleared after the NP-40 alternative was supplemented with cOmplete. In a lysis buffer with 1 M UM171, theprotein concentration was quantified. Supernatants were immunoprecipitated overnight at 4 °C with 25 µl of Pierce anti-HA magnetic beads (Thermo Fisher). Beads were washed six times with lysis buffer, eluted in SDS–PAGE loading buffer and carried forward to immunoblotting as described above.
HDAC1 and CoREST are comprised of full-length HDAC1, which is also referred to as UniProt. The HDAC2–CoREST complex consisting of HDAC2 (chloroform 2– 488) and CoREST (chloroform 86–485) were in ExpiSf9 cells. The human HD AC1 and HDAC2 were cloned using the pFastBac donorVector and the baculoviruses were constructed using the Bac-to-Bac protocol and reagents. The HDAC1 construct was tagged on the C terminus with a FLAG tag, the HDAC2 (amino acids 2–488) construct was tagged on the N terminus with a SUMO tag, which can be cleaved in insect cells and with 6×His on the C terminus. The CoREST was tagged with a 10. His tag followed by an MBP tag on the N terminus. The MIER2 is found in W172K F188C, and six lysines were altered to make it better suited for CoREST. F191E. V197A V201N F209K). Baculoviruses can be prepared according to standard protocols, and these plasmids were used to prepare them. The ExpiSf9 cells were grown before they were ready for expression. For the HDAC1/2–CoREST complex or HDAC1/2 alone expression, the ExpiSf9 cells were either co-infected with HDAC1 or HDAC2 and CoREST baculovirus, or infected with HDAC1/2 baculovirus alone. The cells were incubated for 72 h (26 °C), collected and then frozen with liquid nitrogen for future purification. Cells were resuspended in lysis buffer (50 mM Tris-HCl is cold with a temperature of 300 mM. NaCl is 5 mM. MgCl, 15% glycerol, 1 mM TCEP, 20 mM imidazole) supplemented with 1 mM PMSF, 10 µM leupeptin, 0.5 µM aproptinin and 1 µM pepstatin A and sonicated. Clarifying lysate was done by centrifugation at 100,000g and then incubated with either a nickel affinity or anti-FLAG M2 affinity gel. Resin was washed with lysis buffer, followed by elution using lysis buffer with 200 mM imidazole or 200 µg ml−1 FLAG peptide. The prepacked anion exchange column and the Superdex 200 10/300 GL column are used to get rid of the contaminants. The purity of the recombinant protein was verified by SDS–PAGE and fractions with 90–95% purity were pooled and stored at −80 °C.
The fluorescein labelling of the LSD1– CoREST–HDAC1 complex is now ready for use. A Cys point mutagenesis has been conducted next to the TEV protease cleavage site of N-terminally truncated CoREST for the ligation reaction with NHS-fluorescein54. The reaction buffer was kept at room temperature for 4h after the 2 mM NHS-fluorescein was cultured with 500 mM emesna. The LSD1–CoREST–HDAC1 complex purified by FLAG M2 affinity gel was washed with reaction buffer and incubated with TEV protease for 5 h at 4 °C. The 500 l mixed with the fluorescein/MESNA solution made a final concentration of 125 mM MESNA. The mixture was kept in the dark for 48 hours. The complex was then desalted and further purified by a Superose 6 10/300 column. Fluorescein labelling efficiency was analysed by SDS–PAGE and fluorescence gel imaging (Amersham Typhoon FLA 9500, Cytiva). The purity of the complex was verified by SDS–PAGE and fractions with 90–95% purity were pooled and supplemented with 5% glycerol and stored at −80 °C.
TR-FRET measurements in white microtitre plates (Corning, catalogue no. 3572) and low-volume 384-well plates for ubiquitination assays
Unless otherwise noted, experiments were performed in white, 384-well microtitre plates (Corning, catalogue no. 3572) in 30-μl assay volume, or white, 384-well low-volume microtitre plates (PerkinElmer, catalogue no. 6008280). The following settings were used for TR-FRET measurements on a Tecan plate reader: 340-50-nanesse, 490/10-nanesse, and 520/10-nanesse. The 490/10-nm and 520/10-nm emission channels were acquired with a 50% mirror and a dichroic 510 mirror, respectively, using independently optimized detector gain settings unless specified otherwise. On a per-well basis, the TR-FRET ratio was used as the intensity ratio.
In a one-to-one mixture of ligand buffer, WTKBD4 was diluted to 15 M. NaCl, 1 mM There is a buffer of 20 mM HEPES and TCEP, 10% glycerol. HDAC1– Co-REST, HDAC2–co-rest, HDAC1, HDAC2 are all contained in TCEP. This was aliquoted in triplicate into a black 384-well plate (Corning), followed by twofold serial dilution in assay buffer containing 10 nM JL1 with or without 20 nM recombinant LHC, HDAC1-CoREST, HDAC2-CoREST, HDAC1, HDAC2 or LSD1–CoREST (final volume, 25 µl). The plate was read with the SoftMax Pro software at 1,700 ms integration after being cultured at room temperature for one hour. Wells containing only assay buffer were used for background subtraction. The G-factor was adjusted to set the polarization of assay buffer with 10 nM JL1 and 200 nM LHC only to a reference value of 27 mP. Curves were fitted to the sigmoidal, 4PL model in GraphPad Prism 9.
The ubiquitination assays were set up the same way as before. Reactions were performed at 37 °C in a total volume of 20 µl. The reactions contained 5 mM ATP and 100 M WT ubiquitin. WT KBTBD4 (unless otherwise indicated), 10 µM UM171/DMSO with 25 mM Tris-HCl (pH 7.5), 20 mM 10 M InsP6 and 2.5 mM MgMDL2 is a reaction buffer. Substrate fluorescein–LHC was preincubated with nothing else except the reaction mixture and E1 at 37 C for 5 minutes before starting the reaction. The reactions were quenched by adding a buffer containing a reducing agent. The reaction samples were analysed using the western blotting and blue staining techniques.
Deletions, 1-amino acid substitutions, 3- and 4-terminal substitution of adjacent genes, and 2- and 3-terminal substitution of adjacent genes have been included in the library. The 5′ and 3′ homology arms were added as well as forward and reverse barcodes for the different sub pools of mutations for downstream cloning. The final library was ordered from Twist Biosciences and had a range of single-stranded oligos. The Twist pool was resuspended in Tris-EDTA toconcentration of 1 ng l1 and then the sub pools were separated using a template from the Twist pool. Each pool was amplified with a lsPCR2 primer, and the libraries were gel-rind. The cloned olugos were ordered from the Twist pool and not from the sigma-Aldrich pool.
We used a number of tools to analyse the data, including python, biopython and pandas. When reading a library from unsorted as well as sorted cells, raw reads matching sequence in the library were counted. Counts were then processed by converting them to reads per million, adding a pseudocount of 1 and transforming them by log2. The amount of each variant in GFP+ and GFP populations is quantified by subtracting the log 2-transformed counts for unsorted cells and averaged across replicates. Heatmaps were generated using matplotlib (v.3.7.1).
Position probability matrices of the GFP+ and unsorted populations were constructed for each mutually exclusive category (single substitution, single insertion, double substitution and double insertion) by normalizing raw counts by the total read counts of each corresponding category, averaging across replicates and tallying the probability of every amino acid at each position. The information content, IC, of each position N was calculated according to Kullback–Leibler divergence, which is as follows:
The position probability matrix of the unsorted population was used as a background frequenciesBN. Logos were generated using Logomaker (v.0.8)56.
Sensitivity of tumour cell suspensions to the HDAC1/2 inhibitor RBC1HI – I. MB PDXs harbouring WT or KBTBD4-PR mutant
MB PDXs harbouring WT KBTBD4 (RCMB28 n = 3, MED411FH n = 2) or KBTBD4-PR mutant (ICB1572 n = 5) were used to assess sensitivity to the HDAC1/2 inhibitor RBC1HI. In brief, freshly resected PDX tumours were cut into small pieces, incubated for 30 min at 37 °C in papain solution (10 units per millilitre, Worthington, catalogue no. LS003126) containing N-acetyl-l-cysteine (160 μg ml−1, Sigma-Aldrich, catalogue no. A9165) and DNase I (12 μg ml−1, Sigma-Aldrich, catalogue no. Surrounded by gentle pipetting, DN25 was separated from single cells. Red blood cells in the tumour cell suspension were removed by incubating in RBC Lysis buffer (STEMCELL technologies, catalogue no. 07850) at 37 °C for 2 min, followed by rinsing in DPBS-BSA. Cells were filtered using a 40-µm strainer and counted, and viability assessed to be above 80%. Cells were plated at 1,000 cells per well in 384-well plates in Stem cell media. Serially diluted RBC1HI was immediately added at a final concentration of 40–0.006 µM to the plated cells, with DMSO as negative control, and incubated for 72 h. Cell viability at the end of incubation was measured using Cell Titer-Glo Luminescent Cell Viability Assay 2.0 (Promega) with PHERAstar FSX microplate reader. Raw values were converted to cell viabilities and data analysed using Prism 10 to generate dose–response curves and obtain half-maximum inhibitory concentration values57.
In order to assemble the complex, isolated MBbd4 and UM171 were mixed together, and applied to the Superose6. TCEP (Tris(2-carboxyethyl)phosphine). The isolated complex was quenched with 1 M Tris-HCl pH 8.0 at room temperature. The cross-linked sample was snap-frozen for future use.
To prepare grids for cryo-EM data collection, a QuantiFoil Au R0.6/1 grid (Electron Microscopy Sciences) was glow discharged for 30 s at 20 mA with a glow discharge cleaning system (PELCO easiGlow). 3.0 μl of the purified KBTBD4-UM171-LHC complex at 0.7 mg ml−1 was applied to a freshly glow-discharged grid. The grids were prepared after being put in a chamber at 10 C and 100% relative humidity and then frozen in liquid ethane with the help of the Vitrobot Mark IV system. Data collection was performed on the FEI Titan Glacios transmission electron microscope (Thermo Fisher Scientific) operated at 200 kV at the Arnold and Mabel Beckman Cryo-EM Center of the University of Washington. The automation scheme was implemented using the SerialEM72 software (v.4.1.8) using beam-image shift73 at a nominal magnification of ×105,000, resulting a physical pixel size of 0.885 Å. The images were acquired on a K3 camera direct detector. The total dose was 50 electrons per 2 for each image, and it was fractionated into 99 electron-event representation frames. The data was collected in four sessions. The videos were collected from a wide variety of sources.
11,263 videos were collected and imported into the structure, followed by patch motion correction and patch estimation. In total, 10,057 micrographs were retained after filtering the micrographs with CTF parameters and manual inspection. The blob picker job in CryoSPARC was able to pick over one million particles. A total of 147,826 particles was used for primary ab initio reconstruction, from which the templates were generated, and template picker was conducted to pick 8,280,266 particles. 341,735 particles were selected after two rounds of cleaning by 2D classification. By the end of the second round of cleaning, 766,539 particles were used for reconstruction. After one extra round of cleaning up by heterogenous refinement, 572,349 particles from good reconstruction were selected to get re-extracted without Fourier cropping. The overall resolution was 3.83 with the help of the Heterogeneity and Non-uniform refinement75. Further details about the data processing are provided in Extended Data Fig. 5.
Synthesis of enriched K–GG peptides for the formation of Cilantro 2 eGFP-IRES-m Cherry reporter
The K–GG peptides were enriched using the UbiFast method. For each sample, 500 µg peptides was reconstituted in 250 µl HS bind buffer (Cell Signaling Technology) with 0.01% CHAPS. Reconstituted peptide was added to 5 µl PBS-washed HS anti-K-ε-GG antibody bead slurry (Cell Signaling Technology, 59322) in a 96-well KingFisher plate (Thermo Fisher Scientific). The plate was covered with foil and incubated for 1 h at 4 °C with end-over-end rotation. The King Fisher Flex was used for processing the plate containing the anti-K–GG beads. In brief, bead-bound enriched peptides were washed with 50% acetonitrile/50% HS wash buffer followed by awash in PBS. The K-GG peptides were labelled with 400 g TMTpro reagents for 40 minutes and quenched with 2% hydroxylamine. The beads were then washed with HS wash buffer before being deposited into 100 µl PBS. The supernatant was removed and the enriched K–GG peptide was eluted from the beads. The eluate was desalted using C18 StageTips, frozen and dried in a vacuum centrifuge.
In order to make the MIER1 and RCOR2 inserts into a Cilantro 2 eGFP-IRES-mCherry reporter, they were subjected to a golden-gate assembly. The point alterations were introduced via the standard site directed mutagenesis techniques. The deletion constructs were made using the New England Biolabs and cloned into the Cilantro 2 code using a piece of cloning technology. The particles that were used to transduce the cells were derived from the Cilantro 2 vector. After 48 h, the cells were selected with 1 g muliple puromycin for 5 days. The selected cells were treated with various concentrations of UM171 or 0.1% DMSO for 6 or 24 h. The geometric mean of the ratio of GFP to mCherry fluorescence was calculated for each sample using the NovoExpress software (v.1.5.0, Agilent). The drug-treated samples were normalized to the ratios of the sample that was used for DMSO and plotted using GraphPad Prism. All degradation assays were done in triplicate and FACS-gating schemes are shown in Supplementary Fig. 1a.
Source: UM171 glues asymmetric CRL3–HDAC1/2 assembly to degrade CoREST corepressors
Microscopical Studies of ” A_rm T+B_” Fmoc-Thr(OtBu)-Glyclic Acid using a NanoTemper BLUE System
MST assays were performed with the Monolith NT.115 (NanoTemper) system using the Nano BLUE mode. The exciting laser power was set at 50% and MST power was set to medium. KD values were calculated using MO.analysis (v.2.3) software with the quadratic equation binding KD model shown below:
The issue is ” A_rmT+B_”
Fmoc-Thr(OtBu)- glycolic acid was made using a reported two-step protocol. Then, H3K9ac(1–34) with a sequence as ARTKQTARKS-TGGKAPRKQL-ATKAARKSAP-A-TOG-G was synthesized by standard solid-phase peptide synthesis and purified by reversed-phase HPLC. The Fmoc-protected Amino Acids were purchased from Novabiochem. F40 sortase was expressed and purified as reported previously, and bacterial expression and purification of Xenopus laevis globular H3 (gH3; amino acids 34–135 C110A) were performed also according to a previous protocol61. Next, the F40-sortase-catalysed histone H3 ligation reaction was carried out between the H3K9ac (amino acids 1–34; note that the C-terminal residue is extruded) peptide and the gH3. The pure histone H3K9ac was obtained from the reaction mixture through ion-exchange chromatography.
The methods used for the nucleosome reassembly63 have been reported before. Bacterial expression and purification of X. laevis core histones H2A, H2B and H4 were then carried out, followed by assembly of the histone octamer and refolding as previously reported64. The pre- purification process involved the use of the Supermax 200 10/300 GL column by GE healthcare, which resulted in the purification of the Octamer with a size-exclusion chromatography. The final mixture was subjected to HPLC purification, as well as TEKgel DEAE ION-exchange column to purify the final product. The nucleosome was analysed using TBE-gel, followed by Western blotting and then anti-H3K9ac antibodies.
Source: UM171 glues asymmetric CRL3–HDAC1/2 assembly to degrade CoREST corepressors
Dosage and kinetics of the recombinant HDAC1, HDAC2 and MAZ1600 HDAC substrate28 on microtitre plates for HEK293T cell transduction
Recombinant HDAC1 (BPS Bioscience 50051) or HDAC2 (BPS Bioscience 50002) were diluted to 6 nM (1.2×) into buffer containing 50 mM HEPES, pH 7.5, 100 mM KCl, 0.5 mg ml−1 BSA, 0.001% Tween-20 and 25 μl added to wells of a white, 384-well microtitre plate (Corning 3572). Test compounds were added in serial dilution (1:2 titration, 15-point, cmax = 10 μM) using a D300 digital dispenser (Hewlett-Packard), and allowed to equilibrate for 1 h at room temperature. Then, 5 μl of 6× MAZ1600 HDAC substrate28 was added (final HDAC1/2 concentration 5 nM; final MAZ1600 concentration 18 μM) and deacetylase activity was allowed to proceed for 45 min at room temperature. 5 l of developer solution was added. LBH589 final concentrations) and the plate was incubated for 30 min at room temperature. 7-Amino-4-methyl coumarin fluorescence was measured on the Tecan Spark plate reader: 350/20 nm excitation, 460/10 nm emission. The background was defined by the 10 M LBH589 dose, and the ceiling was defined using a no-inhibitor control. A four-parameter dose–response curve was calculated using background-corrected, normalized, and Prism 9 data.
sgRNAs enriched in the base editing screens were ordered as synthetic oligonucleotides (Azenta/Genewiz), annealed, and ligated into either pRDA_478 or pRDA_479. The plasmids were transfected into HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Then, 48 h after transduction, cells were selected with 2 µg ml−1 puromycin (Thermo Fisher Scientific) for 3 days, then sorted for single-cell clones on the BD FACSAria Cell Sorter (BD Biosciences). Single-cell clones were validated by genotyping and the stability of mutants was assessed by immunoblotting. sgRNA sequences and annotations, as well as primer sequences used for genotyping are provided in Supplementary Tables 3 and 4, respectively.
To assess statistical significance of the resulting clusters, we simulated a null model of random sgRNA enrichment scores. The sgRNA positions were kept fixed and the sgRNA enrichment scores were shuffled around randomly and per-residue. Empirical P values were calculated for each amino acid by comparing its observed resistance score to the null distribution of random resistance scores. The FDR was controlled using the Benjamini–Hochberg procedure. Finally, linear clusters were called by identifying all contiguous intervals of amino acids with adjusted P ≤ 0.05. P values were increased by log10-transformed and divided by 1.