Evaluating the efficacy of totipotency activation of CRISPRa experiments in human ES cells using UMAP and MAPQ data
Low-quality bases and sequenced reads were removed. The clean reads were aligned to both the reference genome and in-house database with Hisat2. Each embryo had its expression level normalized by the TPM. The KD information for each embryo at the 8cell and blastocyst stage as metadata was incorporated together with the gene-expression matrix in Seurat analysis. The individual embryos were visualized by UMAP with default parameters. The TF activity for cells with TE or ICM candidate gene KD was evaluated using SCENIC71.
To evaluate the efficacy of totipotency activation of our CRISPRa experiments in mouse ES cells, we first quantified the abundance of designed sgRNAs targeting candidate TFs (CEBPG, LBX1, ETS2, MEF2D, ESR2, ESR1 and ALX1), positive control TFs (ZSCAN4 and DUX), and non-targeting control based on detected sgRNA unique molecular identifiers (UMIs). sgRNAs that had fewer than 16 UMIs were excluded from further analyses. The cells were projected in UMAP along with totipotent blastomere like cells and pluripotent stem cells. To analyse the perturbation effects of candidate TF genes, we ranked the genes and calculated perturbation correlation among of them based on cells receiving only one sgRNA using MUSIC70 with default parameters. Furthermore, the totipotency score for cells with each combination of gene perturbation was calculated on the basis of the totipotent gene signature.
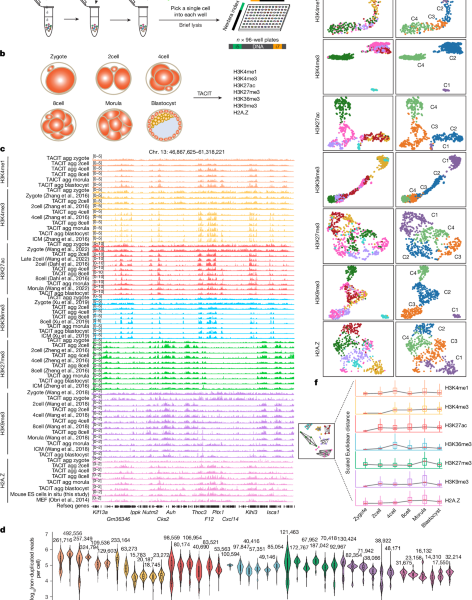
TACIT data were processed as previously described19, but with a few modifications for single cells. Raw TACIT sequencing data were evaluated using FastQC (v.0.11.5), followed by mapping to the mouse reference genome mm10 by Bowtie2 (v.2.2.9)55. Mapped reads with MAPQ vales less than 30 were considered as multi-mapped reads and filtered out using Samtools (v.1.9). It was possible to remove all of the duplicate craps using Picard. For aggregated analysis, single-cell.bam files were merged. The peaks were called with the help of the ‘–broad’ parameters in the macs2 program. Raw CoTACIT sequencing data were de-multiplexed and paired using an in-house code as previously described19. Sequencing data for each histone modification was performed according to the analysis pipeline as described for TACIT data.
The late 2cell stage allvalidPairs matrix was downloaded from the GEO database. To identify interactions, we used the analyzeHiC function of Homer (v.4.11)68 at 50 kb resolution and plotted interactions with Python (v.3.9.7).
We integrated H3K4me1, H3K4me3, H3K36me3 and H3K27ac TACIT profiles with gene expression. In brief, the cell–peak or cell–bin matrix for each histone modification was first generated using cisTopic58. The GeneActivity function of Seurat (v.4) was used to create a gene-activity score matrix based on the cell–peak or cell–bin matrix. Next, anchors between the two modalities were identified with the FindTransferAnchors function. In particular, many titrations were performed to obtain the highest prediction score, including using the cell–peak or cell–bin matrix, or the bin size of the cell–bin matrix. TACIT cells with a prediction score lower than 0.2 were filtered out. Notably, for integrating cells in the 2cell stage, histone-modification signals in non-canonical broad binding regions were excluded before Seurat integration (5 kb for H3K4me3 and H3K27ac, 20 kb for H3K36me3 and H3K4me1).
In Extended Data Fig. 10b, for each chromatin-state labelling, after TF–IDF normalization, SVD dimensionality reduction and cluster finding (as described above), we used the FindMakers function in Seurat (v.4) to find differential bins between cells at the 2cell and 8cell stage. To quantify the establishment of totipotency-related regions, we calculated the percentage of differential bins that had already been annotated as corresponding chromatin states for each synthetic cell.
For clustering cells on the basis of scChromHMM-defined chromatin-state annotations of all genomic intervals (Fig. 4a), we used the posterior probability matrix for each state as input for TF–IDF normalization, SVD dimensionality reduction, cluster finding and UMAP visualization with Seurat (v.4). We used 1:5 dimensions for clustering and visualization. For clustering cells on the basis of chromatin-states annotations in all TSSs, we selected genomic intervals that were ±2 kb flanking TSS regions and averaged the posterior probability for a specific chromatin state in each TSS as the average probability of this chromatin state. Next, we used a mean matrix for each state to find clusters and UMAP visualization. The dimensions were used for visualization.
To integrate the six histone modification profiles, we used the multivariate HMM introduced in ChromHMM61. The.bam files for each synthetic cell were binized with the binarizeBam function. The default parameters were used to learn 12 states on each synthetic cell. To reduce noise and mitochondrial interference, all reads from mitochondrial DNA are filtered out. Next, we annotated the states in three steps: first, we filter out chromatin states with extremely low genome coverage because of technical noise, then we define hidden chromatin states using the combination of histone modifications, and finally, we assign a new state to each state. Finally, we labelled the 12 states as multivalent (all histone modifications), weak promoters (H3K4me3), strong promoters (H3K4me3 and H3K27ac), weak enhancers (H3K4me1), strong enhancers (H3K4me1 and H3K27ac), poised gene bodies (H3K36me3 and repressive histone modifications), active gene bodies (H3K36me3 and active histone modifications), polycomb-protein-associated heterochromatin (only H3K27me3), H3K9me3-associated heterochromatin (only H3K9me3), heterochromatin (H3K27me3 and H3K9me3), and quiescent/low.
We performed hierarchical clustering with 155 RNA synthetic cells after we obtained them with six histone-modification profiles. The number of clusters closely corresponded to the exact cell number of each developmental stage, such as two clusters for the 2cell stage, four clusters for the 4cell stage, and so on. We took the histone-modification profiles of all cells in the same cluster and created 90 synthetic single cells, which included 6 histone modifications. To reduce effects from sequencing depth, we normalized cell numbers and non-duplicated reads before aggregating data.
We integrated the H3K27ac profiles with the Gene expression the same way that was done in step 2. As H3K27ac, H3K27me3 and H3K9me3 profiles were experimentally linked, we directly transferred corresponding H3K27me3 and H3K9me3 profiles to the linked RNA synthetic cells.
We merged five scRNA-seq cells along pseudo time into a single synthetic cell.
The first peaks for aggregated.bam files were used to calculate the genome coverage at each stage. We used the parameters of nomodel and nolambda to call peaks. Next, we binned the mm10 genome into 200-bp genomic intervals, and for each histone modification, genome coverage at a specific stage was calculated as the percentage of genome intervals that overlapped with peaks at that stage. To evaluate genome coverage for single cells, the genome was first binned into 200 bp and bins with histone modification signals ≥ 1 were defined as covered bins. The percentage of covered bins was defined as genome coverage for each single cell.
The multi BigwigSummary function is used to calculate the normalized mean scores in 5-kb bins of the genome. The plot correlation function is used to calculate the Spearman correlation or Pearson correlation.
Incubation and incubation of C57BL/6J female oocytes with sperm suspensions after the hCG injection
The oocytes were collected from female C57BL/6J mice after the hCG injection. Oocytes were incubated in a 200 μl drop of HTF (M1135, Aibei) for 30 min before addition of the sperm suspension. Sperm samples were collected from C57BL/6J male mice and capacitated by placing in a 37 °C, 5% CO2 incubator for 60 min. Next, 3–5 μl of the sperm suspension taken from the edge of the sperm capacitation drop was added to the oocyte clutches (final sperm concentration of 1–5 × 105 cells per ml) and incubated for 3–4 h at 37 °C with 5% CO2. Forcefully pipetting the oocytes up and down several times in a 10 μl volume using a 200 μl pipette helped remove excess sperm. Viable fertilized oocytes were washed and transferred to a new 35 mm culture dish containing KSOM medium (M1435, Aibei). The embryos were distributed evenly throughout the culture dish and incubated at 37 °C with 5% CO2 overnight. The early 2cell and late 2cell embryos were collected after the procedure.
Next, cells were incubated with specific antibody in 100 μl antibody buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine (Sigma), 2 mM EDTA, 0.01% digitonin, 0.05% TX-100, 1% BSA–PBS, 1× cocktail, 10 mM sodium butyrate and 1 mM PMSF) at 4 °C for 3–4 h. Cells were washed twice with a buffer of 20 mM HEPES and 150 mM. NaCl, 0.5 mM spermidine (Sigma), 0.01% digitonin, 0.05% TX-100, 1× cocktail, 10 mM sodium butyrate and 1 mM PMSF) and suspended with 100 μl high-salt Dig-wash buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine (Sigma), 0.01% digitonin, 0.05% TX-100, 1× cocktail, 10 mM sodium butyrate and 1 mM PMSF) containing 3 μg ml–1 PAT–MEA/B. The PAT expression, purification and assembly procedures were performed as per previously described guidelines19. The cells were washed twice with the high-salt buffer and then suspended with a 10 l cold reaction buffer. Adding 10 l 40 mM stopped the reaction. EDTA and cells were washed twice with 1% BSA–PBS, and single cells were picked and placed into a well of a 96-well plate with a mouth pipette under a microscope. The plates were pre-rinsed with 1% BSA–PBS to avoid loss of DNA fragments. Tris-HCl was added to each well. Each well had samples covered with a small amount of mineral oil and placed in a 55 C petri dish for 15 minutes to release DNA fragments. Next, l over 10 mM. To quench the reaction, 1 l of 0.9% Triton X-100 was added to each well. Finally a 17 l mix. The KAPA High-GC buffer, 5 mM dNTP mix and 10.8 l H2O were added to each well. Nextera i5 index primer and 0.5 μl 10 mM i7 index primer (Supplementary Table 1). PCR enrichment was performed in a thermal cycler with the following program: 1 cycle of 72 °C for 5 min; 1 cycle of 95 °C for 3 min; 11 cycles of 98 °C for 20 s, 65 °C for 30 s, 72 °C for 1 min; 1 cycle of 72 °C for 5 min; and hold at 4 °C. The library was selected with 500–1,000 bp fragments and the beads were used to purify it. The beads are from AMPure. The libraries were sequenced with paired-end 150-bp reads on a NovaSeq 6000 platform (Illumina).
For siRNA knockdown, isolated zygotes were microinjected with sets of three siRNAs against targets (20 μM in total) or with non-target control (NC, 20 μM in total). The following siRNAs were used: NC, UGGGACUUGCAGGCCUGAUAUTT; Nanog, CGAGAACUAUUCUUGCUUATT, CCUGAGCUAUAAGCAGGUUAATT and UGGAGUAUCCCAGCAUCCAUUTT; Zfx, GGUUCAUGAUAGUGUAGUATT, GGAUGAAGAUGGACUUGAATT and GGAGGACAACGAAAUGAAATT; Yy2, GCUGCGAGAAGAUGUUCAATT, CACCAUGUGGGACGAUGUUAATT and GACCUAUAGCAUGCUCUCAUATT; Tcf12, GUGGCAGUCAUCCUUAGUCUATT, GAUGCAAUGUCCUUCUUAATT and GGAACAAGUGGUCAACCAATT; Cebpb, GAGCGACGAGUACAAGAUGTT, CACCCUGCGGAACUUGUUCAATT and CGCCUUUAGACCCAUGGAAGUTT; Bbx, UGGGACUUGCAGGCCUGAUAUTT, CCAGUGGGAGCAAGAAGUUUATT and CUCCCUCAAUAUAGUCCUAUUTT; Smad2, GUGAUAGUGCAAUCUUUGUTT, UGGUGUUCAAUCGCAUACUAUTT and CCUUCAGUGCGAUGCUCAATT; Hbp1, CCCUACCCAAUCUGCCAUAUATT, GGCUAACAGAGUUAGCAAATT and CCAGCUAAGUUCAGAUGUATT; Cdx2, GGACAGAAGAUGAGUGGAATT, GAGAAGGAGUUUCACUUUATT and GCUUGCUGCAGACGCUCAATT; Klf6, GCUUGCUGCAGACGCUCAATT, GACCAAUAGCCUGAACUCUTT and GAUGAGUUGACCAGACACUTT; Sox15, CCUGGCAGUUACACCUCUUCTT, GAUGAAGAGAAGCGACCCUUTT and GACUCUUCCACUCCAUAUAAUTT; Med1, UAAGCUUGUGCGUCAAGUAAUTT, GGCUCUCCAAUCCUUAGAACATTand GUGGCCUAUAACACUCUAAUUTT; Elf5, GCCCUGAGAUACUACUAUAAATT, GGACCGAUCUGUUCAGCAATT and GGAGGUUAGUGUACAAAUUTT; and Hif1A, CCAUGUGACCAUGAGGAAATT, GCAGACCCAGUUACAGAAATT and GCAGGAAUUGGAACAUUAUTT. siRNAs were ordered from Hippobio. The injected embryos were transferred to KSOMaa medium (Millipore) and droplets were covered with mineral oil (Sigma) in a Petri dish (Ibidi) and cultured in a tissue incubator (37 °C and 5% CO2) (Thermo Fisher Scientific). A single embryo was collected at the 8 cell stage and a singleembryo was stained to confirm the KD or marker gene expression.
The following antibodies were used for TACIT (catalogue and lot numbers provided after the supplier name): H3K4me1 (1:50; Abcam, ab8895, GR3369516-1); H3K4me3 (1:200; Millipore, 04-745, 3243412); H3K27ac (1:500; Diagenode, C15410196, A1723-0041D); H3K36me3 (1:200; Active Motif, 61101, 06221007); H3K27me3 (1:200; Millipore, 07-449, 3146226); H3K9me3 (1:200; Active Motif, 39161, 30220003); and H2A.Z (1:200, Abcam, ab4174, GR279096-1). Donkey anti-rabbit-Alexa 488 (1:500; Invitrogen, A32790) and donkey anti-rabbit-Alexa 555 (1:500, Invitrogen, A31572) were used as secondary antibodies. Antibodies used in immunofluorescence staining included SOX2 (1:200; Active Motif, 39843, 2226414) and CDX2 (1:200, BioGenex, MU392A-UC, MU392A0516D).
Wild-type V6.5 mouse ES cells were cultured at 37 °C with 5% CO2 and were maintained on 0.1% gelatin-coated plates in high-glucose DMEM culture medium containing 15% fetal bovine serum (Invitrogen), 1% penicillin–streptomycin (Hyclone), 1% MEM nonessential amino acids (Cellgro), 1% Glutamax (Gibco), 1% nucleoside (Millipore), 0.1 mM 2-mercaptoethanol (Sigma) and 1,000 U ml–1 recombinant leukaemia inhibitory factor (Millipore).
Source: Genome-coverage single-cell histone modifications for embryo lineage tracing
Fear Conditioning Training of C57BL/6 Mice in M2 Medium: A Random Walk in the Follies and Long Walk With a Pre-Warmed Folly
To make zygotes, a mass of them surrounded by the cells of the Follies was placed into a 1 hyaluronidase solution and held at 37 C for a few minutes. The zygotes were then transferred to M2 medium and their zona pellucida was gently removed by treating with pre-warmed Tyrode’s acidic solution (Sigma) for several minutes. The second polar bodies were manually removed by a very fine glass needle.
Aged C57BL/6 mice (16–21 months old) were obtained from the National Institute on Aging rodent colony. Young C57BL/6 mice (3 months old) were obtained from Jackson Laboratories. Male mice were used in the experiments. All mice were kept on a 12 h–12 h light–dark cycle and provided ad libitum access to food and water. All animal care and procedures complied with the Animal Welfare Act and were in accordance with institutional guidelines and approved by the Veterans Affairs Palo Alto Committee on Animal Research and the institutional administrative panel of laboratory animal care at Stanford University.
Data handling and analyses were done using R studio and GraphPad. All statistical analyses comparing measurements between two groups were carried out using unpaired two-tailed Student’s t-tests. All statistical analyses comparing measurements between three or more groups were carried out using one-way ANOVA tests with post hoc tests for multiple comparisons. A P value of <0.05 was considered significant. All procedures involving multiple groups were done in alternating fashion, to avoid temporal and technical biases. Data in Figs. There are 1b–d, h–o, 2a, b,f–j, 3e–n, 4f–i and 5j–l extended data figs. In at least two independent experiments, 2a, 3f, 7a,h,i and 8h-k were successfully replicated.
In fear conditioning tests, mice were trained to associate cage context and an audiovisual cue with an aversive stimulus (foot shock). On the first day of training, mice were placed in a cage with 30 s of cue light and a 1000-hertz tone followed by a foot shock with a 180-s interval. On the second day, mice were re-exposed to the same cage context, and freezing behaviour was recorded during minutes 1.5 to 6 using a FreezeScan tracking system. After 2 min of exploration, mice were exposed to the same light and tone from the day before in a novel context, but with different smell, floor texture, and chamber walls. Freezing behaviour was recorded for 1.5–6 min following the cue using the FreezeScan tracking system (Cleversys).
In the Y maze test, the Y maze is made up of 3 white, opaque plastic arms. At the beginning of the trials, mice were placed in the end of 1 arm and allowed to freely explore all 3 arms for 5 min. An arm entry was defined as having all four limbs inside an arm. The maze was cleaned with 70% ethanol between animals and before the first animal to eliminate traces of odour. The numbers of arm entries and triads were recorded. The number of triads was used to divide the number of possible changes by 100.
Pseudobulk counts and differential gene expression in M. musculus studied using SoupX (v4.1.1) and DESeq2
Pseudobulk counts were derived by aggregating raw counts for each sample using Seurat’s AggregateExpression function. DESeq2 was then used to perform bulk data normalization and differential gene expression across groups using default parameters. Each comparison was carried out using the Benjamini–Hochberg procedure. Genes with FDR <0.05 were used for subsequent KEGG pathway enrichment and Metascape43 analysis (Supplementary Data 5).
Gene counts were obtained by aligning reads to the M. musculus reference genome GRCm38 using CellRanger software (v4.0.0) (10X Genomics). Ambient RNA was removed from each sample using SoupX (v1.6.2) and droplets containing multiple nuclei were filtered out using DoubletFinder (v2.0.4). We used Seurat (v4.1.1) to further exclude cells with fewer than 200 or more than 5,000 features and cells with more than 10% mitochondrial genes. There were 69, 250 nuclei that remained, which were used for more analysis. 3e–d.
Pooled cortical and Hippocampal tissue were removed from frozen brain hemispheres of young and older mice that had been injected with an AAV 8 weeks prior to the event. The brain tissue was prepared with a razor blade and then transferred to a glass Dounce tissue grinder with two grams of lysis buffer on ice. The tissues were collected using 25 strokes with pestle A and 25 strokes with pestle B. After 5 min of ice on ice, a 500g sample was put in a freezer for 5 min and then placed into a bottle at 4C. The pellet was resuspended with 4 ml EZ lysis buffer, incubated on ice for another 5 min, and pelleted as before. The pellet was resuspended with chilled PBS and then blocked with a mouse Fc block in the FACS buffer. Nuclei were subsequently stained with AF647-anti-NeuN (1:100, Abcam, ab190565) in FACS buffer with 0.2 U µl−1 RNase inhibitor (Takara, 2313 A) for 30 min on ice, washed with FACS buffer, and resuspended in FACS buffer with Hoechst dye (1:2,000, Thermo Fisher, H3570) and 0.2 U µl−1 RNase inhibitor (Takara, 2313 A). Over 50,000 singlet nuclei were sorted into 1.5 m Eppendorf tubes with 10 Mgml UltraPure BSA and 0.2 U l1 RNase inhibitors in them. The nuclei were 400g for 5 minutes at 4 C. Supernatant was removed leaving about 40 µl of suspended nuclei. The chondriei were counted using a haemocytometer and assessed for concentration and quality. snRNA-seq libraries were prepared using the Chromium Single Cell 3′ The manufacturer has a specific plan for targeting 10,000 nuclei per sample. The cDNA was generated using a mixture of 12 PCR cycles and 11 in the final generation library. The quality control of libraries and cDNA was done using a High Sensitivity D5000 ScreenTape. The libraries were read on a NovaSeq X.
For production of PHP.V1-sCLDN5::EGFP and all other plasmids containing the PHP.V1 (pUCmini-iCAP-PHP.V1 was a gift from V. Gradinaru; Addgene plasmid #127847) capsid, AAV production was performed in-house utilizing a previously published protocol42. There was triple transfection of HeK289T cells done on the 90 to 95 percent of confluent cells. The fresh medium was replaced 12 h after thefection. Medium was collected 72 h post-transfection. Fresh, warm medium was added and collected along with cells 120 h post-transfection and combined with the previous fraction. The cells and medium were kept at room temperature for 15 minutes. Supernatant was collected in a separate bottle and combined with 40%. When the final concentration is 8% wt andvol, it’s ready to be put on ice for two hours before it reaches 4 C. After the cell pellet was resuspended in a buffer containing salt- active nuclease, it was put into a petri dish at 37 C and then transferred to 4 C overnight. PEG medium was centrifuged at 4,000g for 30 min at 4 °C. After centrifugation, the supernatant was thrown away. PEG pellet was resuspended in SAN + SAN buffer, combined with the previous fraction, and incubated at 37 °C for an additional 30 min. Supernatant was loaded onto the iodixanol gradient after lysis was performed for 15 minutes at room temperature. There were two h and 25 minutes for the transfer to the ultracentrifuge, which was set at 350,000g. The AAV particles were washed in PBS and then concentrated using an Amicon Ultra-15 filter device. AAV titration was performed using the AAVpro Titration Kit (for Real Time PCR) Ver.2 (Takara Bio). AAVs were injected retro-orbitally at 8 × 1011 viral genomes per mouse.
The number is 26. MiniPromoter (‘sCLDN5’) pEMS1938 was a gift from E. Simpson (Addgene plasmid #82563). A cis rAAV genome plasmid with AAV2 inverted terminal repeats was utilized for cloning of a sCLDN5 and EGFP reporter using restriction enzymes and In-Fusion Snap Assembly (Takara Bio). To knock down C1galt1 in brain endothelial cells, de novo predictions of small interfering RNA (siRNA) guides targeting C1galt1 were generated using the DSIR algorithm39 and subsequently filtered using ‘Sensor rules’ to select for sequences with highly favourable small hairpin RNA (shRNA) features40,41. Three de novo 97-mer miR-E shRNA sequences (Supplementary Table 2) were synthesized (IDT) and inserted into pAAV-sCLDN5-EGFP using restriction enzyme cloning for in vitro evaluation. C1GALT1 and B3G NT3 were cloned into pAAV-s CCLDN5-EGFP using restriction enzyme cloning to create pA.
Brain microvessels were isolated and lysed in a buffer on ice. Lysates were centrifuged at 13,000g for 15 min at 4 °C, and supernatant protein concentration was measured by microBCA (Pierce). Samples were then reduced and alkylated by TCEP and CAA, followed by protein purification using SP3 (Cytiva). Samples were digested with 200 ng trypsin/LysC in pH 7.8 at 37 °C for 3 h, desalted, and eluted following the manufacturer’s instructions. Before going onto the system, samples were acidified to 1% formic acid. All samples were injected with a single microgram Protein. The LC–MS/MS analysis was performed on the Tims TOF Pro with the solvent A and solvent B. Tryptic Peptides were loaded first on the trapping column Water’s ACQUITY UPLC M-Class Symmetry C18 Trap Column, 100 A, 5m, 180 m, 20mm, and eluted with analytical column. Elution gradient was set as 0 min 5% B; 9 min 12% B; 9.1 min 12% B; 27 min 30% B; 27.5 min 85% B and 36 min 85% B with a flow rate of 0.35 μl min−1 from 0 min to 9 min and reduced to 0.3 μl min−1 at 9.1 min until the end of the gradient. The measured edites were measured with a base method of m/z range 100–1,700 and 1/k0 range of 0.85–1.30Vs cm2. The source parameters were 1,400 V for capillary voltage, 3.0 l min−1 for dry gas, and 180 °C for dry temperature using Captive Spray (Bruker Daltonics). The total collision energies were allocated for 1/ K0 and 1/K0 respectively. Data were processed using Spectronaut (Biognosys AG, v19.1) for directDIA search with Swiss-Prot Mouse database downloaded on 3 March 2023. Default settings were used with a slight modification of minimum peptide length 6. Candidates were sorted by Q 0.05 and absolute average log2 ratio.
For flow cytometry analysis, bEnd.3 cells were plated in a 24-well plate and treated with 5 nM StcE for 16 h at 37 °C. Cells were collected from plates using enzyme-free cell dissociation buffer (Thermo Fisher Scientific, 1315014) and resuspended in 1% BSA in PBS (FACS buffer). The cells were stained with anti-CD54, FITC-anti-VCAM1, and lysates of chlorpyrifos by using ice in FACS. Live cells were identified using Sytox Blue viability dye (1:1,000, Thermo Fisher Scientific, S34857). Data was analysed using the FlowJo software and analysis was performed on a Sony SH800S sorter. Live singlet cells were used for analysis.
The mouse brain endothelial cell line bEnd.3 was cultured in a humidified incubator and supplemented with 10% fetal bovine sera and 1% penicillin/strepomycin. For bulk RNA-seq analysis, bEnd.3 cells were grown in 6-well plates and treated with 5 nM StcE for 16 h at 37 °C. The RNeasy Plus Micro kit allows the lysing of cells and collection of the total RNA in the Eppendorf tubes. RNA quantity and quality were assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies). All samples passed a high quality control threshold and went to cDNA library preparation. Libraries were sequenced on the NovaSeq 6000 (paired-end, 2× 150 bp depth). Trimmed reads were close to the M. musculus reference genome. DESeq2 was used to analyze and view differential gene expression. GO enrichment analysis used genes with a Padj 0.05.
There are previously published evidence of brain cell enrichment, which were chosen for analysis. We looked for genes listed in the KEGG that were related to glycosylation. Most glycoproteins were excluded due to the enormous variety of members in this family which possess biological functions not directly relevant to glycosylation. The threshold of significant enrichment was set at Padj 0.05 and the genes were used for reactionome pathway analysis.
LC–MS/MS analysis was performed on a Q Exactive HF-X (Thermo Fisher Scientific) with an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). 40 cm of reproSil- Pur 120 C18-AQ 1.9 m was packed in an in-house 75-m capillary column. The separation took place using a flow rate of 300 Nested units per second with a 120 min gradient, which included 98% A + 4% B for 18 min, 80% A + 30% B for 72 min, and 40% B for 15 min. The scanned objects were acquired at a resolution of 60,000 and the maximum injection time was 20 ms, and the range was 300–1,650 m/z. MS2 scans were acquired with the following parameters: resolution of 15,000, AGC target of 1 × 105, maximum IT of 54 ms, loop count 15, TopN 15, isolation window 1.4 m/z, fixed first mass 100.0 m/z, normalized collision energy (NCE) 28 units, charge exclusion of unassigned, 1, 6–8 and >8, peptide match preferred, exclude isotopes on, and fragmented m/z values were dynamically excluded from further selection for a period of 45 s. Raw data were processed and analysed using MaxQuant and Perseus36. A target-decoy Mus musculus reference proteome database was downloaded from Uniprot. Variable alterations were specified such as methamphetamine oxidation and N-terminal acetylation. Precursor ion search tolerance of 20 ppm and product ion mass tolerance of 20 ppm were used for searches. Both unique and razor peptides were used for quantification. Results were filtered to a 1% false discovery rate (FDR) at the peptide and protein levels. MAXLFQ37 had a minimum ratio of 1 and was used to quantifigate and normalized the genes. For quantitative comparative analysis, protein intensity values were log2-transformed, and missing values were imputed from a normal distribution with width 0.3 and downshift value of 1.8 using Perseus. The principal component analysis was performed in Perseus using the Benjamini–Hochberg FDR. DAVID38 was used as a background for GO term enrichments.
The human immune monitoring center at theSTANFORD University has a luminex test. The mouse was used according to instructions from the manufacturer. Standard curve and quality control samples were also used along with theDiluted 1:3 samples. All wells were added to the test to assess nonspecific binding.
All recombinant StcE proteins were produced as described22,23. For all applications, proteins were run through Pierce high-capacity endotoxin removal columns (Thermo Fisher Scientific) at least seven times following manufacturer’s instructions. Endotoxin levels were tested using HEK-Blue lipopolysaccharide (LPS) Detection Kit 2 (InvivoGen) according to manufacturer recommendations. The mice were injected in the middle of the day and given ice-cold PBS to last them for 2 days. Cerebral bleeding could be visualized by using the eye post-perfusion and H&E staining. Hemibrains were cut into 5m-thick sections and put into slides for H&E staining. Sections were deparaffinized in xylene three times, hydrated in a series of graded alcohols, and then stained with Richard Allan haematoxylin. Sections were then dehydrated (10 dips in 95% ethanol followed by 2× 1 min in 100% ethanol), cleared in xylene (3 times, 1 min), and coverslipped prior to imaging on a wide-field microscope (Zeiss AxioImager).
The mice were given 0.25 g1 body weight with Sulfo-NHS-biotin. The tracer was allowed to circulate for 5 min before perfusion with PBS. Hemibrains were post-fixed in 4% PFA overnight at 4 °C, cryopreserved in 30% sucrose, and sagittally sectioned into 40-μm slices. The sections were blocked with CD31 and the appropriate secondary antibodies as was described earlier, in addition to being co-stained. Images were taken on a confocal laser-scanning microscope (Zeiss LSM880) and analysed using ImageJ software. Multicoloured gradient images were generated using the fire LUT in ImageJ. Permeability index of vessels was determined as the area occupied by tracer divided by the vessel area.
The mice were euthanized with 2.5%. You can get avertin and transrCardially perfused by using ice cold solutions such as 10 g and PBS. Mice were euthanized with 2% (v/v) for all other studies. Unless noted otherwise, avertin and manually perfused with PBS. Tissues were extracted and fixed in 4% PFA at 4 °C overnight before preservation in 30% sucrose in PBS. The tissue was sectioned into 40 m slices using a microtome. The slices were initially blocked in donkey blood with 3% normal, then 3% normal, and then 3% normal with Triton X-100 at room temperature for 1.5 h, and then 4% normal with the following primary antibodies: goat anti-CD31 (13:100,000), R&D anti-CD31 (1 The following day, slices were washed three times with TBS-T, stained with the appropriate Alexa Fluor-conjugated secondary antibodies (1:250, Thermo Fisher Scientific) or Alexa Fluor-conjugated streptavidin (1:1,000, Thermo Fisher Scientific) for 2 h at room temperature, washed three times again, mounted, and coverslipped with Vectashield Hardset Antifade Mounting Medium with DAPI (Vector Labs, H-1500-10). Images were analysed using ImageJ after being seen on a confocal laser-scanning microscope. The luminal blood vessels were considered to be the marker of interest divided by the total vessel area. Endothelial MFI calculated using CD31+ mask.
The fresh-frozen brain tissues were obtained after the death of a loved one and received approval from the Institutional Review Board. All samples used in this study were stored at 80 C until the time of processing, and the autopsy was done after 12 h. Supplementary Data 2 has group characteristics summarized. People in the Alzheimer’s disease group have been diagnosed and pathologically determined to have Alzheimer’s brain disease hallmarks.
For BBB ultrastructural analysis, mice were injected retro-orbitally with 0.3 ml of 0.5 mg g−1 of HRP type II in PBS (Sigma, P8250). The brains were fixed in a 0.1 M cacodylate buffer and then at room temperature for one hour and then at 4 for 16 hours. After washing overnight with 0.1 M sodium cacodylate, the tissues were cut into 1mm-thick sections using a matrix. Cortical punches (0.5–1 mm3) were cut and incubated in 0.5 mg ml−1 of 3,3′-diaminobenzidine with 0.01% hydrogen peroxide in TBS for 45 min at room temperature. Tissues were washed with TBS overnight, post-fixed in 2% osmium tetroxide and 2.5% potassium ferrocyanide in 0.1 M sodium cacodylate, and en bloc stained with 1% uranyl acetate and Walton’s lead aspartate stain. Samples were then dehydrated in an ascending ethanol gradient and embedded in epoxy resin (EMS). Eighty-nanometre sections were cut using a Leica UC7 ultramicrotome and collected on formvar-coated copper grids. The grids were post-stained with 3.5% uranyl acetate followed by Sato’s lead citrate. Sections were imaged using a Tecnai 12 120 kV TEM (FEI), and data were recorded using a Rio16 CMOS camera with GWS software (Gatan). The assessment of tight junctions in the images was performed in a blinded manner.

